Rare diseases: Phelan-Mcdermid Syndrome
Last February 28 was held the day World of Rare Diseases. The syndrome of Phelan-Mcdermid form part of the list of rare diseases that affect in Spain for 3 million people.
On February 29, 2008 was held for the first time the World Day of Rare Diseases. This commemoration is carried out in various places around the world and is usually performed on February 29 (if it is not a leap year). During this day various activities are held to give greater visibility to the rare diseases, those that affect a small proportion of the population.
However, in each country the “rare diseases” have different legal definitions. In Europe , for example, is considered a rare disease that affects 1 in every 2,000 people. In the United States to that suffered by fewer than 200,000 people, while in Japan to which it affects less than 50,000 . Although in Italy, there is no legal definition of a “rare disease”, is regarded as rare disease to one that affects from 1 of every 20 thousand inhabitants up to 1 of every 200 thousand inhabitants. Most (80 %) of the rare diseases are genetic, while others are the result of infections and allergies, or due to degenerative causes and proliferating.
The European Organization for Rare Diseases (Eurordis) estimates there are between 5,000 7,000 different rare diseases, and that these have a prevalence (the number of people living with a disease in a given time) of between 6% and 8% of the population in the European Union, i.e. affect between 44 and 59 million Europeans. Phelan-Mc Dermid Syndrome syndrome deletion of 22q13.3 (Phelan-Mc Dermid syndrome, PMS) is a neurodevelopmental disorder characterized by a delay in the development, normal or accelerated growth.
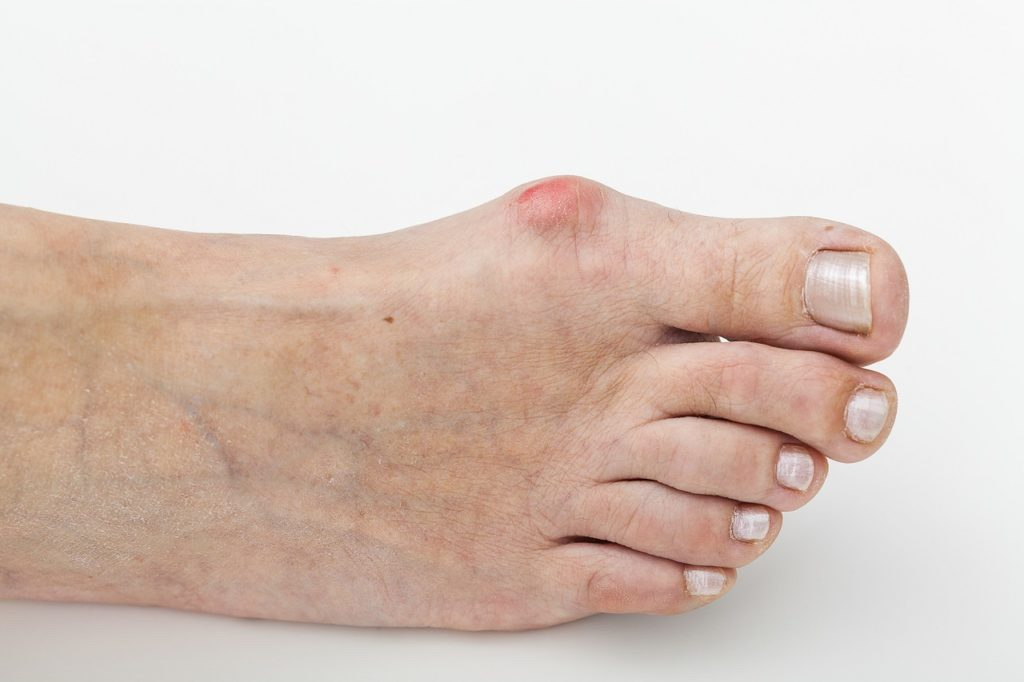
Most of the affected children have a cognitive disability (more or less serious) in learning and language. Presented facial dysmorphism minors, such as toenails thin and brittle (78 % ); large hands (68 % ); big feet; prominent ears and not very well formed (65 % ); and other features that are not obvious to the visual examination as high tolerance to pain (86 % ); seizures (percentage unknown); abnormalities of the spinal cord; poor central vision, etc. The hypotonia, feeding problems, and developmental delay are often the earliest symptoms to identify in this syndrome. Some of the patients with Phelan-Mc Dermid exhibit certain autism spectrum disorders, although recent research has concluded that the behavior of children with TDC does not fit within the criteria of classical autism.
The margin of the main clinical features these patients may suffer from other clinical manifestations or pathologies, which in addition to the time of consultation with neurologists, endocrine, geneticists, may require additional specialized care with other specialists, as well as other additional tests, which sometimes may not be implemented due to a lack of resources or ignorance. Certain associated problems such as seizures, breathing or heart problems are addressed with conventional treatments. The ability to communicate in these patients is quite determinant for its ability to be autonomous. The adaptive sports, music therapy and sensory integration increase the awareness of the child.
Causes of the Phelan-Mcdermid Syndrom (PMS)
The PMS is caused mainly by different deletions in size (loss of genetic material) in the chromosomal region 22q13.3. To a lesser extent can also occur as a result of an unbalanced translocation between chromosomes (one of them being the chromosome 22), by the formation of a ring chromosome (when stabilized by the loss of genetic material)or investment that affect the long arm of chromosome 22 or by point mutations (one to several nucleotides) affecting a single gene, the gene SHANK3. The protein encoded by this gene plays an important role in the maturation and stabilization of synapses between neurons in the brain. The loss of one copy of the gene SHANK3 seems to be the main cause of the deletion 22q13.
For the moment, we cannot establish the existence of other genetic or environmental factors important in the pathogenesis of this syndrome. In most cases it is a spontaneous mutation, de novo, where the deletion affects the terminal region of the long arm of chromosome 22 (the paternal chromosome in 75% of cases). Although, there is also a hereditary form due to chromosomal translocations involving the family chromosome 22. Different studies to date seem to indicate that there is no clear relation, except for some clinical feature, between the size of the deletion and the greater or lesser severity of the clinical spectrum in this pathology.
Although, recent information seems to indicate the existence of a serious neurological deterioration, retrogression and the bipolar disorder in adolescent and adult patients, suggesting that the PMS fenotipo can evolve with the age. Up to the date, the clinical bogey in PMS has been generally studied deeply in the childhood and the evolution of the fenotipo in adult age has researched in very few cases. Longitudinal remarks it will help the doctors to construct a model of the most useful syndrome clinically, allowing that the early treatment and an appropriate care of the patient. For it, it is important to have big cohorts of these patients. The Institute of Medical and Molecular Genetics (INGEMM) of the University Hospital La Paz has in collaboration with different Spanish clinical centers, and the Association of PMS Spain of a cohort concerning the hundred of patients.
We do not know the predominance of the syndrome of microdeleción 22q13.3 in Spain, probably it is infra – diagnosed so much clinically, as at laboratory level. There exists an international record of patients of PMS, in which there collaborates the Association PMS Spain.
In general, in recent works a frequency of 0,28 % has been estimated in 15.000 children with intellectual disability, autism and/or congenital defects. And the fact is that from the genetic point of view, this anomaly often is not visible in the cariotipo and to establish its diagnosis it demands skills of molecular citogenética, like the FISH (hybridization for fluorescence in Situ) or skills of molecular biology like MLPA (Multiplex ligand dependent Probe Amplification). On the other hand there is the arrival of the microarrays, based on the hybridization genómica compared (CGH array: aCGH). There are a relatively new skill that is used for the analysis of the genome in search of profit and losses of chromosomal material. This method has a resolution and a clinical yield major than technical of classic citogenética (10-1000 times).
Investigation and results
As we were commenting previously, the hope in many of these rare illnesses happens for the component of the fundamental research before being able to move to the clinical investigation and hence to the clinical assistance. The investigation lines on SHANK3, the gene responsible for this syndrome, have allowed to developcellular and animal models that promise new treatments for the syndrome of Phelan Mc-Dermid and for the autism. In fact, The evidence of preclinical studies in the mouse and human models neuronales with shortcoming in SHANK3 suggest that IGF-1 (insulin-likegrowth factor 1), the plasticity is capable of revertir sináptica and the motive learning deficits in a chosen group of patients with PMS.
Finally, individuals with the syndrome of Phelan McDermid must receive medical attention of routine of its doctor of primary health care. Nevertheless, the review and multidisciplinary pursuit for a clinical geneticist, neuropsiquiatras in specializing centers will help to improve its quality of life. This way for example, the physical and occupational therapy and the exercise help them to improve the coordination and the force. Also it can be suitable to apply strategies that improve the communication, treatments of orthodontics for the bad dental occlusion and medicines to treat the hyperactivity, anxiety, etc. In this sense there is promoted the consolidation of certain initial multidisciplinary consultations of evaluation to characterize systematically, with genetic tests and of neuroimagen, etc.
The periodic evaluations will be necessary to update the professionals of the evolution of the patients and on the other hand to the parents to receive new information about this syndrome. The creation of an association of fond patients, like the Association PMS Spain, allows to the families to give itself mutual support, share its knowledge on the illness, the prevention and the treatments of the same one and to make it come to the rest of citizens.
Dr Julián Nevado Blanco, PhD, MBA
Responsable de Genómica Estructural y Funcional
INGEMM (Instituto de Genética Médica y Molecular). Hospital Universitario la Paz
Investigador IdiPaz y CIBERER.
Referencias:
1. Kolevzon A, Bush L, Wang AT, Halpern D, Frank Y, Grodberg D, Rapaport R, Tavassoli T, Chaplin W, Soorya
L, Buxbaum JD. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid
syndrome. Mol Autism. 2014 Dec 12;5(1):54. doi: 10.1186/2040-2392-5-54.
2.- Phelan K, McDermid HE (2012) The 22q13.3 deletion syndrome (Phelan-McDermid syndrome).
MolSyndromol2, 186-201
3.- Phelan K, Betancur C (2011) Clinical utility gene card for: deletion 22q13 syndrome. Eur J Hum Genet 19
4.- Sarasua SM et al. (2011) Association between deletion size and important phenotypes expands the genomic
region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome). J Med Genet 48, 761-766
5.-Dhar SU et al. (2010) 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH.
Páginas Web:
https://pmsiregistry.patientcrossroads.org
www.omim.org
Post a Comment